
|

|

|

|
| 図1a 茶葉を煮出します。 | 図1b 冷やすと澱が出てきて茶色く濁る。 | 図1c 炭酸ナトリウムを加えると澱が溶けて、 どす黒い、あまり飲みたくない感じの液体になる。 | 図1d 2本の遠沈管に分け取り、それぞれジクロロメタンを 1 mL ずつ加えて、シャカシャカ振り混ぜます。 |
テキストでは茶葉からカフェインを抽出することにしていますが、 各自の興味で、コーヒーや目覚ましドリンクなど、 挑戦してもいいことにしています。 ただし甘味料の入ったものなどだと、 後の操作でシロップ状になることがあり、 避けるのが無難です。 それでも挑戦する若者、無論、大歓迎です。
まずはティーバッグに水を入れて煮出し、 カフェインを抽出します (以前は炭酸ナトリウム溶液で煮出す設定にしていましたが、 ちょっと趣きがないので、お茶を淹れる設定にしました)。 この煮出し液を冷やすと、澱(おり)が出てきて茶色く濁ります (図1b。アイスティーを自作する時によく出会います。 その道ではクリームダウン cream down というそうです)。 これはタンニン tannin が沈殿してきているのです (というか、カフェインなどのアルカロイドと塩を作って沈殿する酸性物質をタンニンと呼びます。 最近は「ポリフェノール」と呼ばれるようです)。 ここに炭酸ナトリウムを加えアルカリ性にすると、 塩を作っていたタンニンは溶解して、 図1c のようなこげ茶色の溶液になり、カフェインなども溶け出してきます。
えられたこげ茶色の溶液を2本の遠沈管(15 mL)にほぼ同量ずつ分け取り、 それぞれにジクロロメタンを 1 mL ぐらい取って蓋をし、 シャカシャカ振り混ぜます (ジクロロメタンは蒸気圧が大きいので、 そのままスポイトで取るとスポイト内で蒸発して、 取ったジクロロメタンが吹き出してきたりします。 一旦、ジクロロメタン瓶の中でスポイトを空打ちして、 ジクロロメタンの蒸気をスポイトに吸い込んでから操作するようにするとよいでしょう)。 こうしたお手軽な分液操作では、遠沈管は便利です。 がんばり過ぎると、懸濁液(エマルジョン)になって分離しにくくなりますが、 そんなに気にしなくても大丈夫です。
|
|
|
|
|
| 図1a 茶葉を煮出します。 | 図1b 冷やすと澱が出てきて茶色く濁る。 | 図1c 炭酸ナトリウムを加えると澱が溶けて、 どす黒い、あまり飲みたくない感じの液体になる。 | 図1d 2本の遠沈管に分け取り、それぞれジクロロメタンを 1 mL ずつ加えて、シャカシャカ振り混ぜます。 |
ジクロロメタンを加えて振盪した液は、 そのままではなかなか分離してこないので、 遠心分離器にかけて分離します。 最初の頃は液相分離ろ紙 (シリコンで処理して水をはじき、有機溶媒とよく濡れるようにしたろ紙。 有機層がろ液として出てくる) を使用していたのですが、 ろ紙に吸収される分、溶媒がたくさん必要になり、 また溶液が炭酸ナトリウムで塩基性が高いことから長時間おくと水層が出てきたりして、 遠心分離機を使うようにしました。 遠心管を使っているので、 このあたりスムーズです。
遠心分離して沈んできたジクロロメタン層は、 スポイトで回収します。 ちょっとぐらい水層が混ざっても構いません。 1度ジクロロメタンでの抽出が終わったら、 もう一度、ジクロロメタンを 1 mL 加えて同様に抽出、 遠心分離して抽出液を得て、先の抽出液と合わせます。 カフェインは水にかなり溶け(室温付近で水に質量分率で 2 % ぐらいまで溶ける)、 ジクロロメタンへの分配係数は 10 ぐらいなものです (酢酸エチルだと分配係数は 1 ぐらいで、回収率はかなり落ちる)。 液量の比を考えると、気分的には1回の抽出で 50 %、 2回の抽出で 75 % 回収できることになります。
抽出液には水層が入っていたりするので、 無水硫酸ナトリウムで乾燥させます (ジクロロメタンへの水の溶解度は 20 °C で質量比 0.15 %で、 溶解している水の量自体は無視できる程度。ただし水へのジクロロメタンの溶解度は1ケタ大きく 1.7 %。 抽出済みの液を、活性炭処理ぐらいで安易に流しちゃダメ!)。 硫酸ナトリウムは取りあえず、混入している水層の量より少し多い目ぐらいに加えて振り混ぜ、 その後、様子を見ながら水層が固結するまで加えていくというのがよいでしょう。 なお硫酸ナトリウムによる乾燥では、 十水塩 Na2SO4·10H2O ができるように考えている人がいますが、 十水塩になるにはかなりの時間を要して、 せいぜい30 分程度の乾燥では七水塩ぐらいであることが多いようです。

|

|

|

|
| 図2a 使用している簡易型遠心分離機。 今時、こんな遠心分離機にはまず出会えません。 | 図2b 遠心分離すると、ジクロロメタン層が分離してくる。 | 図2c スポイトで分取したジクロロメタン層。 少々水層が入ってきても構わない。 | 図2d 無水硫酸ナトリウムを加えて脱水。 もう少し硫酸ナトリウムを加えた方がよい。 |
混入した水層などを硫酸ナトリウムを加えて除いた抽出液は、 サンプル管に移してジクロロメタンを揮発させます。 この時、固結した硫酸ナトリウムに付着した抽出液は、 少量のジクロロメタンで洗って回収します。 サンプル管がせいぜい 5 mL ぐらいのサイズなので、 2回に分けて揮発操作をするのがいいでしょう。
揮発操作は局所排気装置の下で、 ホットプレートでサンプル管を加熱しながら、 送風装置(金魚のプクプク)でジクロロメタンの蒸気を飛ばしながら行います。 ジクロロメタンの沸点が 40 °C なので、 天板温度は 40 °C ぐらいにセットしておけば無難です (上げ過ぎて突沸すると厄介)。 ここで直径 16.5 mm のちょっと小さめのサンプル管(業界でいう No. 1、4 mL の規格)を使うのは、 後段の昇華精製にスムーズに移行するためです。 リンの実験でも使用しますが、 ホットプレートの天板上で加熱するのに使うアルミブロックを、 ちょうどこのサイズ用に作ったので合わせています。 抽出液の液量が多い時には、試験管などで揮発操作をし、 得られた粗カフェインをサンプル管に移して作業してもよいでしょう。
得られる粗カフェインは、 ちょっと色のついた、 茶の匂いのする粉です (この段階では、おそらく一水和物 C8H10N4O2·H2O )。 最初にカフェイン飲料などを選ぶと、 シロップ状のものが残ったりしますが、 気にせず先に進みましょう。

|

|

|

|
| 図3a 加熱に使用するアルミブロック | 図3b 金魚のプクプクで空気を送って蒸気を飛ばす | 図3c 得られた粗カフェイン。 匂いを嗅いでみましょう。 | 図3d 以前は湯浴中で揮発操作をしていました。 |
カフェイン(融点 236 °C)は昇華性を示し、 だいたい 150 °C を超えるようになると、 はっきり昇華が検知できるようになります。 けれどもある程度まとまった量をえるには、 200 °C ぐらいには加熱する必要があります (昇華は蒸留と比べると熱伝導が悪いため効率が落ちます)。
昇華に使用する装置は、 装置というのもおこがましいような、 サンプル管に冷却管 cold finger を差し込んだだけのものです。 サンプル管は先の揮発操作の時と同じように、 アルミブロックに差し込んで、ホットプレート上で加熱し、 冷却管には水を入れておきます。 冷却管のサンプル管への固定は、脱脂綿で巻いて、 差し込む程度で十分です。 なおせっかくの機会なので外径 6 mm のガラス管を使って、 ガラス細工で自作してもらうようにしています。
天板の温度をテキストにあるように 200~220 °C ぐらいにして昇華させると、 だいたい10 分もすれば昇華は終わるでしょう。 ここであまり長く頑張ると、 冷却管の中の水が沸騰して飛び出すことがあるので注意します。
なお直接茶葉を加熱しても、 カフェインが昇華してきます。 収量はかなり低いですが、 カフェインの確認には使えます (インスタントコーヒーではうまくいきませんでした)。

|

|

|

|

|
| 図4a ガラス細工をしてもらいます | 図4b 冷却管 cold finger | 図4c こんな感じで昇華精製します | 図4d 加熱終了後。 結晶が付いています | 図4e マイクロスコープで見たカフェインの結晶 |

| 図5 赤外線分光光度計 Cary 630 で赤外吸収スペクトルを測る |
カフェインの同定には、 赤外線吸収スペクトルを用います。 赤外線吸収スペクトルで見るのは、電磁波の波長がおよそ 2 µm ~ 20 µm 、 波数にして 500 cm-1 ~ 5000 cm-1 の領域で、 主に分子振動に由来する吸収になります。 特に有機化合物の場合には、 分子内の種々の原子団(カルボニル基やメチル基など)が、 あたかも独立に存在しているかのように扱える場合が多く、 それがスペクトルに鋭い吸収として反映され、 その分子特有の情報を与えてくれるので、 同定には好都合です。
学生実験では Agilent の赤外線分光光度計 Cary 630 を使用して、 ATR(Attenuated Total Reflection。減衰全反射)法を用いて、 赤外吸収スペクトルを測ります。 この手法では、 表面に存在する物質の吸収スペクトルを、 反射してくる赤外線の強度から求めます。 ですから ATR装置の台(プリズム。ここではダイアモンドの薄膜を使っています)に、 測りたい物質を付着させれば測定の準備完了で、 後は装置に任せればよいのです (試料の量は 10 μg 程度でも可。測定後の試料回収も可能)。 以前の透過光を用いる手法に比べ、 サンプルの作成に要する労力が格段に少なく、 今回のような同定作業での赤外吸収スペクトルの測定などは、 ほとんど ATR 法に置き換えられています。
測定の際には、大きく次の3点に注意してもらえればいいでしょう。
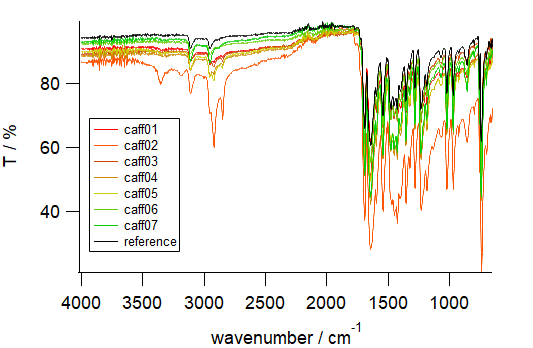
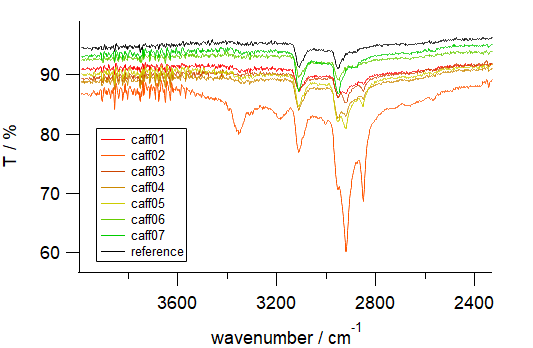
実際に得られた赤外線吸収スペクトルの例を図6 に示します。 caff01~07 の試料すべてについて、 カフェインの標品(無水カフェイン。和研の特級試薬)の吸収が再現されており、 カフェインが含まれていることがわかります。 けれども caff06 と caff07 以外については、 標品にない吸収が現れており、 得られたカフェインに何か入っていたようです。 高波数の 2500~3500 cm-1 に顕著で(図6b)、 2924 cm-1 と 2851 cm-1 の吸収が caff01~ caff05 に認められ、 caff02(橙色)についてはさらに、 3353 cm-1 と 3187 cm-1 にも吸収が見られます (caff02 の 3000 cm-1 付近のなだらかな吸収は水)。
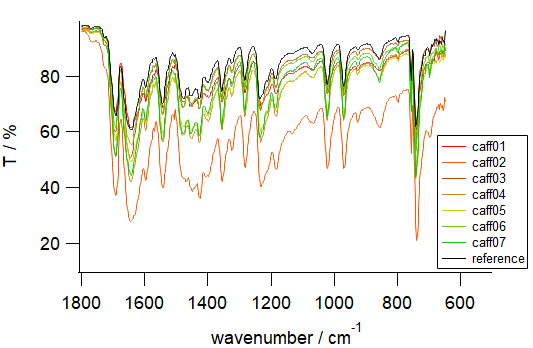
この一方、カルボニル基の吸収の見られる 1800~1500 cm-1 前後や、 いわゆる指紋領域 fingerprint region と呼ばれる 1500 cm-1 程度以下の領域では、 標品にない目だった吸収は現れていません (図6c。caff02(橙色)の1650 cm-1 付近のなだらかな吸収は液体の水と思われます。 同様に3000 cm-1 付近のなだらかな吸収も水)。 こういったところから考えると、 先の2924 cm-1、2851 cm-1 などに見られた吸収はカフェインの何らかの水和物が、 一部できていると見るのがよさそうです。 事前に100 °C ぐらいで乾燥する、 昇華する時に減圧するなどの措置を施こすといいんでしょうか。 まあたいていは標品と一致するスペクトルが得られ、 ムレキシド反応も問題なく出るので、 あまりこだわる必要がない気もしますが・・・
|
|
|
| 図6a 測定全領域。 黒線は無水カフェインの標準サンプル | 図6b 高波数領域。 実験で得られたカフェインの中には、 標品に見られない吸収が存在するケースがある(カフェインの水和物?) | 図6c 指紋領域。 実験で得られたカフェインの吸収は、 標品と一致している (caff02(橙色)については、 試料が湿っていたらしい) |
|
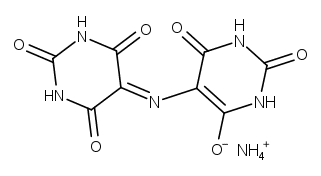
| ムレキシド |
カフェインの ”化学的” な同定法、定性試験として、 ムレキシド反応 murexide test を利用する手法が有名です。
ムレキシドという物質自身は、 19世紀の半ばに染料として市販され (グアノ(鳥の糞の石化したもの。尿酸が含まれる)から作られたらしい)、 古くからよく知られていたようです。 分析化学の分野では、キレート滴定の黎明期、 シュバルツェンバッハ Schwarzenbach が紹介した、 EBT などと並ぶ由緒正しい(?)金属指示薬の一つで、 今も使用されています。
ムレキシド反応は、 カフェイン特有の呈色反応というわけではなく、 尿酸などのプリン塩基を酸化すると、 ムレキシドの類縁化合物が生成して赤く呈色することを利用しています。 ここで採用した手法は、薬局方にあるもので、 過酸化水素で酸化します(薬局方ではテオフィリン(アミノフィリン)の定性にも利用)。 他にも硝酸を使ったり、塩素酸カリウムを使ったりする手法もあるようです。 実験する上での注意点は、 温度を上げ過ぎないことでしょうか。 反応を早く終わらせようと、 天板温度を 200 °C ぐらいまで上げたりすると、 呈色が出なくなります。
なお少し趣向は違っていますが、 アミノ酸のニンヒドリン反応でできる赤色物質(ルーエマン紫 Ruhemann's purple)も、 ムレキシドとよく似た構造を持っています。
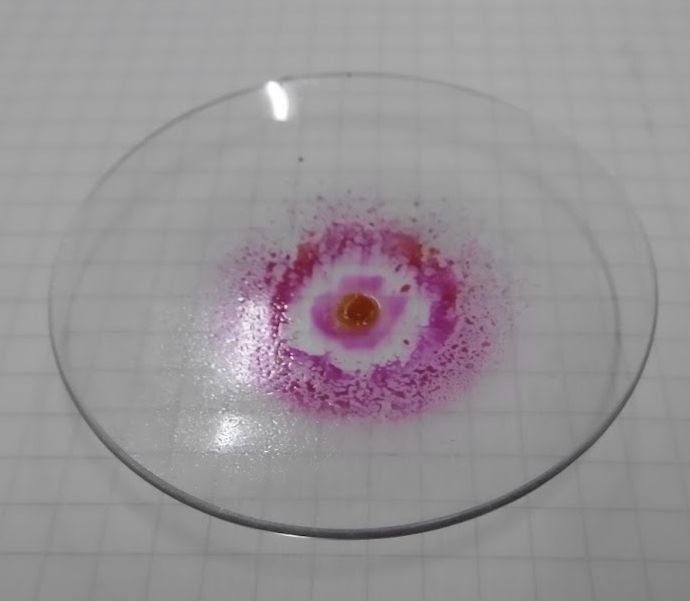
|
|
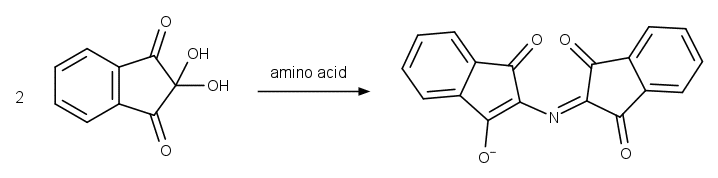
|
| 図7a ムレキシド反応で、最終アンモニアを作用させ赤くなったところ。 | 図7b ヒートガンでブンブン加熱してムレキシド反応をやった例。 ほとんど呈色が見られない | 図7c ニンヒドリン反応。 生成する赤色物質は、ムレキシドとよく似た構造を持つ |