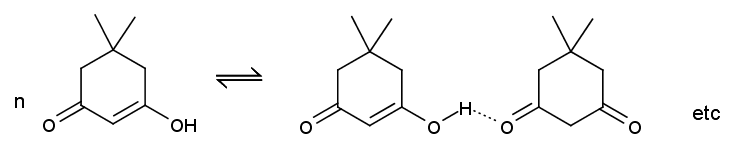
2020.10
吉村洋介
Michael 付加とハロホルム反応
テキストの pdf 版はこちら。
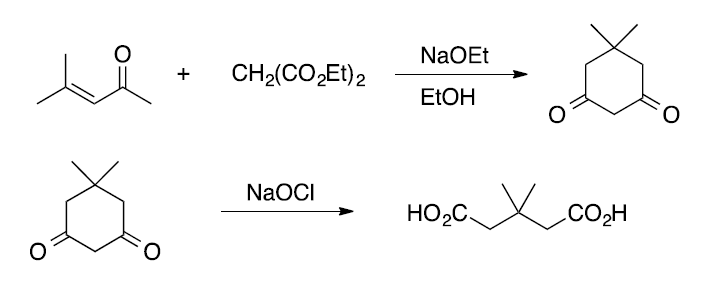
(1)5,5-ジメチル-1,3-シクロヘキサンジオン(5,5-dimethyl-1,3-cyclohexanedione, ジメドン dimedone)
乾燥した100 mL三口フラスコに回転子を入れ、滴下漏斗と還流冷却器(内部が乾燥していることを確認)を付け、
ナトリウムエトキシドの20 %エタノール溶液12 mL(ナトリウムエトキシド30 mmol相当) を加え、
オイルバス中で50 °C 程度に加温する。
そこに攪拌しながらマロン酸ジエチル32 mmolを加え、3 mLのエタノールで希釈した4-メチル-3-ペンテン-2-オン(メシチルオキシド)30 mmolを滴下漏斗からゆっくり加える。
反応溶液をオイルバス中で30分還流した後、温かいまま水酸化ナトリウム75 mmolを水15 mLに溶かした溶液を加え、
さらに1時間半程度還流する。
還流冷却器を外して、反応混合物が熱いうちに4 M塩酸(M = mol/L)をゆっくり加えて中和しpH 2~3程度にする(試験紙で確認。25 mL程度必要。発泡注意)。
発泡がある程度収まったら、蒸留装置を付け、オイルバス中で加熱しアルコールを留去する(約15 mL)。
放冷すると生成物が晶出する。晶出した固体を吸引ろ過後、水で洗い、さらに5 mL程度の氷冷したアセトンで洗った後、乾燥する。
収量2.5 g(60%)。
得られた5,5-ジメチル-1,3-シクロヘキサンジオンは次の合成に使用するには十分純粋だが、NMRスペクトルを取るための試料を得るために再結晶する。
再結晶には0.5 g程度の粗生成物を取り、6倍程度の熱アセトンに溶かした後、ほぼ同量のヘキサンを加え徐冷する。
得られた精製試料はダイアフラムポンプを用いて乾燥し、NMRスペクトルを測定する。
(2) 3,3-ジメチルグルタール酸(3,3-dimethylglutaric acid)
ドラフト中、回転子を入れた100 mLナスフラスコに次亜塩素酸ナトリウム水溶液 を有効塩素 Cl2 にして30 mmol分加え、
撹拌しながら氷浴中で冷却する。これに7.0 mmolの5,5-ジメチル-1,3-シクロヘキサンジオン(ジメドン)を加えた後、
氷冷をやめ約1時間撹拌を続ける。
反応終了後、撹拌しながら15 %亜硫酸ナトリウム溶液7 mL(亜硫酸ナトリウム9 mmol相当)を加え、
残存する次亜塩素酸ナトリウムを分解(還元)し、試験紙でpHが6以上であることを確認する注1。
次亜塩素酸ナトリウムの分解が終わったら、フラスコ底部に沈んだ液体をパスツールピペットでサンプル管に分取し、
IRスペクトルを取ってクロロホルムが含まれていることを確認する。
溶液を分液漏斗に移し15 mLの酢酸エチルで1回洗う。
有機層を分離し、試験紙で確認しながら水層に4 M塩酸を加えて中和してpH 2~3程度にした後、
15 mLの酢酸エチルで3回抽出する。
酢酸エチル溶液を一つにし、無水硫酸ナトリウムで乾燥した後、固体が析出するまでロータリーエバポレーターで有機溶媒を留去する。
残渣を少量の温酢酸エチル(2~3 mL程度)に溶かした後、
ヘキサンを加えて再結晶する。収量0.45 g(40 %)。
(注1)pH試験紙でpHを確認後、1分程度で試験紙が脱色するようなら、未反応の次亜塩素酸ナトリウムが残っているので、亜硫酸ナトリウム溶液を追加する。
ジメドンの合成のこと
ジメドン(5,5-ジメチル-1,3-シクロヘキサンジオン。メソンとも呼ばれる)は、
古くからアルデヒドの分析に利用され、
シックハウスなどで問題になるホルムアルデヒドの分析の公定法にも採用されています
(有害物質を含有する家庭用品の規制に関する法律施行規則。
アセチルアセトンと試料を反応させ発色を見るのですが、
その発色が本当にホルムアルデヒドに由来するかどうか確認するのに、
あらかじめ試料とジメドンを反応させ、発色が変化するかどうかをチェックします。
ジメドンはアルデヒドと 2:1 で反応します)。

摺りの部分にアルカリが付いたままだと、
摺りが取れなくなります。
取り合えず超音波洗浄機に付けて様子を見て、
これで取れない時は・・・

この課題で扱うジメドンの合成反応は、
マイケル付加やクライゼン縮合、βケト酸の脱炭酸といった、
教科書的な反応が盛沢山に詰め込まれています。
またケト-エノール互変異性平衡に関わって、
ジメドンは多彩な表情を見せます。
そうした意味で「教育的」な反応といえるでしょう。
でもその内実は結構厄介です。
この実験ではアルカリを使用して1時間以上還流したりするので、
ガラス器具の摺りの部分には多めにグリースを付けるなりして、
噛みつかないよう予防策を講じておく必要があります。
Grignard 反応で摺りの噛みつきが起きた例はさほどありませんが、
このジメドンの合成ではやかましく言っても、
噛みつきの起きなかったことがありません。
いっそ摺りの部分にシーリングテープ(テフロンテープ)を巻いておいた方がよいかもしれません。
ナトリウムエトキシドとの反応
Grignard 反応の場合のように乾燥器であらかじめ器具を加熱乾燥する必要はありませんが、
例年よくあるのはGrignard の時と同様、ジムロート Dimroth 冷却器の中の水が抜けていないケースです。
水が入ると最初の還流で泥状にならず醤油のような状態になります。
またエタノールは一斗缶からそのまま汲んできたようなのは拙く、
取り合えずモレキュラーシーブで乾燥させておいたものを提供するようにします。
マロン酸ジエチルは滴下ロートから入れ(ここはドボドボ入れて可)、
その後メシチルオキド(4-メチル-3-ペンテン-2-オン)のエタノール希釈液を継ぎ足し、
ゆっくり滴下していく(ここはゆっくり)のが美しいと思います。
最初に50 °C 程度に加温することが指定してあるのは、
加温せず室温でマロン酸ジエチルを加えると、マロン酸ジエチルのナトリウム塩が析出してきてゼリー状になってしまうためです
(昔は金属ナトリウムを使用してナトリウムエトキシドを得ていたので、
自ずと熱溶液になっていたので特に加熱する必要はなかった)。
ここにメシチルオキシドを滴下して還流すると、だんだん沈殿が増え、最後は泥状になります。
今やメシチレン mesitylene (1,3,5-トリメチルベンゼン C9H12)は知っていても、
メシチルオキシドという名前を知らない TA さんがほとんどです。
メシチレンはアセトンと濃硫酸の反応でえられる(3C3H6O → C9H12 + 3H2O)
のですが(その昔、19世紀も前半、アセトンをメシット mesit と呼んでいたらしい)、
この時の副産物として生じることからメシチルオキシドという名前を得たようです
(アセトン2分子のアルドール縮合から得られる。2C3H6O → C6H10O + H2O)。
だいたいがアセトンからメシチレンができる、
酢酸カルシウムの乾留でアセトンが作られていたといった話自体が、
まったく通じない時代になったので、しょうがないのかもしれませんが・・・
なお学生さんのレポートにはほとんど(まったく?)言及されていないのが残念ですが、
マロン酸ジエチルの量(32 mmol)を少しメシチルオキシド(30 mmol)より多く取って、
メシチルオキシドをゆっくり滴下するのは、メシチルオキシド同士のアルドール縮合を防ぐことを期しています。
このあたりは教員、TA諸氏から学生諸君に注意を促してもらうべきかもしれませんが。

|

|

|
|
メシチルオキシドを加えて還流。
数分で濁ってくる。
|
泥状になってくる。
|
どこかの温泉で見るような風景
|
水酸化ナトリウム溶液との反応
水酸化ナトリウム溶液を加えると泥状の物質は溶け、溶液が相分離して2相になり、
これを10分程度加熱還流すると1相になります。
この後さらに1時間半程度還流するわけですが、1時間経過したあたりで沈殿が析出してきます。
原因はよくわかりませんが、この沈殿の析出が起きない場合もあります。
ただし沈殿ができなくても、最後のジメドンの収率にはあまり関係ないみたいです。

|

|

|

|
|
水酸化ナトリウム溶液を加えると、
泥はほとんど溶ける。
最初の内は2相に分離して、
少し濁った感じ。
|
10分ほど還流していると1相になり透明になる。
|
1時間ほどたつと濁ってくる(濁らない場合もある)。
|
だんだん沈殿が増えていく(沈殿ができない場合もある)。
|
脱炭酸とエタノールの留去
塩酸を加えて酸性にしてカルボン酸にして、
脱炭酸反応を起こさせます。
調製した塩酸濃度の精度(たぶん 4 ± 0.4 mol/L ぐらい)にもよりますが 25 mLより小過剰必要になるはずです(計算上NaOHは105 mmol存在します)。
pH が下がって溶液の色が黄色い感じになり、
気体の発生が始まります。
この状態で冷却管を付け加熱蒸留して、
エタノールを留去します。
エタノールが除かれるにつれ、
液が濁ってきて油分が分離してきます。
エタノールが出てこなくなったら加熱を止め、
摺りの部分を外して放冷。
液は酸性になっているのですが、
摺りの部分にアルカリが残っていたりすると噛みつくことがあるので、
念のため摺りの部分を外しておきます。
後は翌日のこと。

|

|

|

|
|
塩酸を加えて pH 2~3 ぐらいにすると濁りが消え、
液は黄色くなり、発泡が起きる。
|
冷却管を付け、発泡が続く中、加熱してエタノールを留去する。
エタノールが減るにつれ、少し濁ってきて油滴が現れるようになる。
|
15 mL ほどエタノールが出たところで蒸留を止め、おいておくと2相に分離する。
冷却管を外し、摺りの部分をほどいて、翌日まで放置。
|
翌日にはこんな感じでジメドンの結晶が出てくる
(油が浮かんだままの時は、最初に水が入ったりした可能性アリ。
酢酸エチルで抽出してエバポレーターにかけたりすると、
収量は低いがジメドンが取れることもある)。
|
結晶の採取と再結晶
フラスコから析出してくるジメドンは黄褐色で、かなり油分が付いています。
次のハロホルム反応の原料にする分には、そのままでも使えますが、
NMRスペクトルなどを詳細に検討するには、精製する必要があります。
そこでスペクトル用に 0.5 gほどを取って、アセトン-ヘキサンから再結晶するようにしています。
再結晶に使用するジメドンは量が少なく、現在使用している直径 70 mmのヌッチェは大きすぎるので、
桐山ロートを使用してもらいます。
桐山ロートの使用法についてTAさんの指導を仰いでください
(いささかお高いものなので取り扱い注意!)。
再結晶後の乾燥は、
量も少なくアセトン-ヘキサン溶媒なので、真空乾燥器を使用する必要はなく、
50 mL ぐらいのナスフラスコに入れ、ドライヤーで加熱しながら、ダイアフラムポンプでしばらく吸引するぐらいでよいでしょう。

|

|
|
ヌッチェで晶出したジメドンを分取。
油分が付いていたりするので5 mL 程度のアセトンで洗浄する。
|
粗生成物(左のカップ)をアセトンから再結晶すると、
白色結晶(右のカップ)となる。
|
ジメドンの合成のはなし
ジメドン (5,5-ジメチル-1,3-シクロヘキサンジオン)-ジメチルグルタール酸の合成の課題は、
手元の実験テキストで見ると1981年以前から実施されていたようで、
ジアセトンアルコール→メシチルオキシド (4-メチル-3-ペンテン-2-オン)→ジメドン→ジメチルグルタール酸という流れで行われていました:
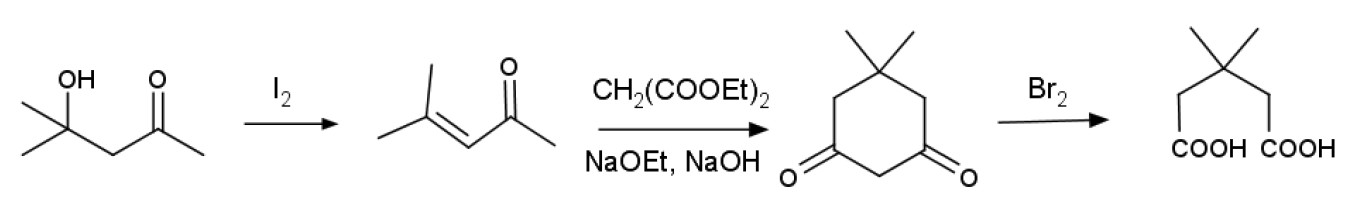
これが2015年度から日程の関係でメシチルオキシドの合成が外され、
メシチルオキシド→ジメドン→ジメチルグルタール酸という形で実施されるような形になったわけです。
(ここでは触れませんが、
メシチルオキシドの合成はOrg. Syn. 1, 53 (1921) を参照して設計されたようです。
個人的には、
相分離・反応系の蒸留操作で、
不思議な色合いの変化があり、
また生成するメシチルオキシドの H-NMR の解釈(シスとトランスのメチル H の化学シフト、
異性体の 4-メチル-4-ペンテン-2-オンの存在)なども興味深かったのですが、
有機系のスタッフには受けが悪く、消えてしまうこととなりました。
当初(1983年まで)のジメドン合成の実験手順は、Org. Syn. Coll. Vol. 2, 200 (1941) 所載のものに準じており、
スケールを1/3にしたものでした
(それでもマロン酸ジエチル 57 g、メシチルオキシド 30 g、金属ナトリウム7.7 g 使用といったスケール)。
またジメチルグルタール酸合成はOrg. Syn. Coll. Vol. 4, p.345 (1963) のもので塩素を臭素に置き換え、
スケールを1/2にしたものに相当しています
(実験テキスト通りであれば臭素を180 g、ジメドンを 75 g使用。
さすがに実際は、これをスケールダウンして実施していたものと推測します)。
この課題についての大きな改定は1998年度で、実験スケールが 1/5 に縮小され
(マロン酸ジエチル 11.4 g、メシチルオキシド 6.6 g。結局 Org. Syn. の 1/10 スケール)、
金属ナトリウムをやめて、
市販の固体のナトリウムエトキシド使用するようになりました。
ぼくが関わった 2019年度の改定では、
実験スケールをさらに小さく半分以下にして、
使用するナトリウムエトキシドを固形物から市販の20 %溶液に切り替えました。
またスペクトル試料用に再結晶操作など加えています。
反応機構のこと
この合成反応の基本的な流れは、
マロン酸エステルのメシチルオキシドへのマイケル付加と、
それに引き続くクライゼン縮合(型)の環化付加反応、
そして脱炭酸ということになります。
実験に照らして、もっともらしい流れを書くと次のようになると思います:
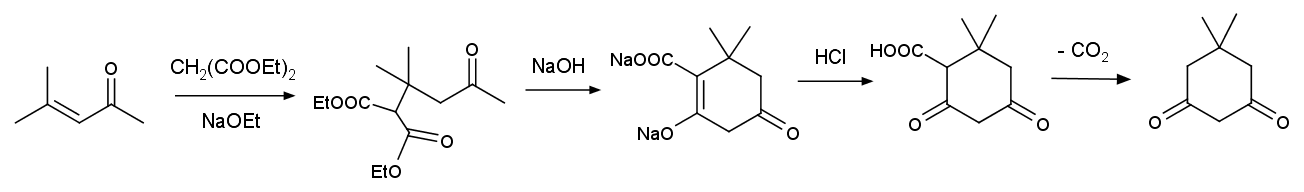
たぶん「常識的」には、環化付加反応が起きるのは、
最初のナトリウムエトキシド NaOEt との反応の段階であると考えるでしょう。
実際、Org. Syn. Coll. Vol. 2, 200 (1941)
の記載もそうなっています。
上記のスキームに基づいて反応機構を書くと、
試験では×になる可能性大です。
確かに「アセチル基のプロトンをエトキシアニオン EtO- が引き抜いてできるカルボアニオンが、
エステルの炭素を攻撃します」
などと言われるとその気になります。
けれどもそれでは、水酸化ナトリウム溶液での処理には、
エステルの加水分解ぐらいの意味しかないことになります。
実験で観察された、最初に2相に分かれて10分程度で1相になったこと、
さらに1時間ぐらい還流して沈殿が生じてきたことがうまく説明できません。
またクライゼン縮合させるには、
ナトリウムエトキシド NaOEt の量が足りません
(単純に考えるとクライゼン縮合までさせるには、
NaOEt はメシチルオキシドの2当量必要なはず)。
所要時間を考えてもエトキシドとの還流時間より、
水酸化ナトリウム溶液を加えてからの還流の方を長くとるのが一般です。
単にエステルの加水分解だけなら、
こんなに長く還流する必要はないでしょう。
Org. Syn. ではエトキシドとの還流時間2時間に対し、
水酸化カリウム溶液
(以前の学生実験でも水酸化ナトリウムではなく水酸化カリウムを使っていました)
を加えての還流6時間。
F. G. Mann and B. C. Saunders, "Practical Organic Chemistry," Longman 1978
(1960年版の復刻版。かつて英国の Cambridge などで広く使われた教科書らしい)
では、エトキシドとの還流時間1時間に対し、
水酸化ナトリウム溶液を加えての還流1.5時間以上という指定になっています。
今回の処方では、時間内に終わらせるべく、
還流時間をいずれも短めに設定してあるのですが、
この水酸化ナトリウム溶液を加えての還流時間は、
あまりケチるわけにいかないところでした。
収率を追求したい向きは、もう少し還流時間を取ってもらうのがよいでしょう。
収率のこと
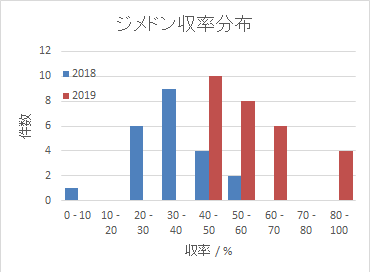
上記テキストには収率を 60 %(2.5 g)と記載したのですが、
これは昨年度実施した際の学生諸君の平均値になります。
右図に示すのは、
2018年度と2019年度の学生諸君のジメドンの収率の分布です。
グループによりばらつきはありますが、
スケール、処方を変更した 2019年度は収率が大きく向上したことがわかります。
2018年度の平均収率 35 %(中央値も35 %)に対し、
2019年度は平均収率 59 %(中央値は58 %)でした。
この収率の”躍進”の要因としては、
スケールが小さくなったことによる操作性の向上、
ナトリウムエトキシド溶液の使用による塩基性度の向上もあるでしょうが、
何より 2018年度までのテキストでは、最終、析出したジメドンを採取するところで
「固体を吸引濾過後、水で洗い、さらに少量の氷冷したアセトンで着色がほぼなくなるまで洗う」
と指示されていたため(現行は 「5 mL」と必要量を明示)、
大量のアセトンで洗浄して収率の低下を招いていたことがあるように思われます。
なお、文献所載の収率は 67 - 85 %(Org. Syn. 再結晶後)、
80 %(Mann & Saunders。これは学生実験用のテキストで本実験同様、粗収率)で、
いずれも 20 ポイントほど当方の平均を上回っています。
2019年度の諸君の結果でも4グループは、84~89 %という飛び抜けた結果を出していました。
直接聞いても見たのですが、
はかり間違いなどではなく、
実際に3.5 g 以上のジメドンを得ていました(それも比較的薄い黄色のきれいな品)。
何か秘密があるのか聞いてみたのですが、
ご当人たちにも思い当たる節がないとのこと。
何か隠された実験のツボがあるかもしれませんが、
今後の検討に待ちたいところです。
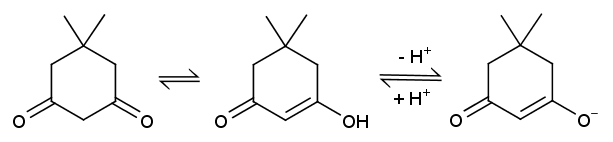
ジメドンのケト-エノール平衡
ジメドンはケト-エノール平衡を示し、
塩化鉄(III)溶液で紫色に呈色します(エノール型の存在)。
また水溶液では pKa が 5.2 程度の炭酸より強い酸として振る舞い、
炭酸水素ナトリウム溶液に二酸化炭素を発生しながら溶けます
(実際に試してみて欲しいところですが、やってみる学生さんがほとんど(まったく?)いないのは残念)。
結晶のジメドン
ジメドンは結晶中ではエノール型として存在し、
X 線結晶構造解析からはエノール型のジメドン分子が、
水素結合でらせん状に連なっているさまが読み取れます。
このことは今回の実験で測定する赤外(IR)吸収スペクトルからも示唆されるところです。
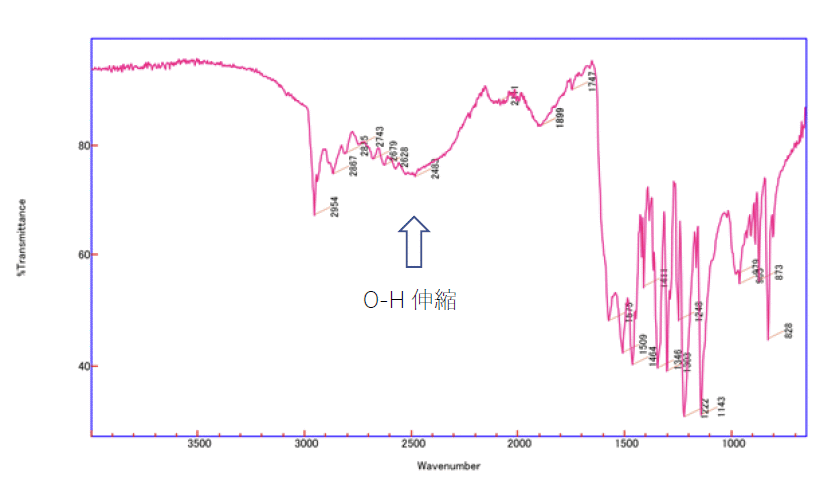
ジメドンの IR スペクトル(ATR 法。Cary 630)。
2300 ~ 3000 cm
-1 ぐらいに幅の広い吸収がある。
IR スペクトルには、2300 ~ 3000 cm-1 ぐらいに幅広い吸収があるのですが、
あまりに巾が広すぎて、見逃してしまう人が多いようです。
同じ O-H の伸縮振動に分類されますが、
トリフェニルメタノールの場合のように 3400 ~ 3500 cm-1 ぐらいに 100 cm-1 の幅(これもかなり広いが)で出ることもあれば、
今回のジメドンの場合のように 2500 cm-1 ぐらいに 数百 cm-1 の幅で出ることもあり、
注意が必要です(安息香酸などカルボン酸もこうしたパターン)。
なおこの IR スペクトルには、カルボニルの吸収に相当する1620 cm-1 ぐらいの吸収(共役カルボニルなので低波数側に来る)が判然とは見えません。
これは測定法、測定機種によるもので、
詳しい話はこちらを参照ください。
溶液中のジメドン
アセチルアセトンとジメドンのケト(K)-エノール(E)平衡の平衡定数 [E]/[K] の溶媒依存性。
20 °C。
C. Reichardt and T. Welton, "Solvents and Solvent Effects in Organic Chemistry," 4th ed. Wiley-VCH 2010 より
| 溶媒 | アセチルアセトン | ジメドン |
| ベンゼン | 14.7 | 0.12 |
| エタノール | 5.8 | 169 |
| 水 | 0.23 | 19 |
| クロロホルム | 5.94 | 0.05 |
| DMSO | 2.0 | 19 |
溶液中でジメドンはケト-エノールの平衡にあり、
この平衡は溶媒・ジメドンの濃度に強く依存することが知られています。
右の表に種々の溶媒中の、アセチルアセトンとジメドンのケト-エノール平衡の平衡定数 [エノール]/[ケト] を示します。
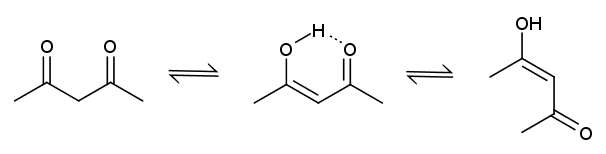
アセチルアセトンはアルコール中でケト型が安定になり、
ジメドンではエノール型が安定になっています。
この原因として挙げられるのは、
アセチルアセトンの場合にはエノール型が分子内水素結合を作って安定化できることです:
ジメドンのエノール型の場合には、立体的にこうした分子内水素結合を作ることができません。
したがって溶媒との水素結合を形成することで、
エノール型の安定化が起きるというシナリオが有効になります。
この時、溶媒に水素結合するのか(ドナー)、されるのか(アクセプター)で話はちがってくるわけですが、
ジメドンのエノール型の安定化には、溶媒の水素結合受容能(アクセプター性)が重要な因子になるようです。
クロロホルムは水素結合する能力(Cl3C-H···)はあるのですが、
水素結合受容能が低く、エノール型があまり安定にならないとして表の結果は理解できます。
さて溶媒の水素結合を受け取る能力が低いとき、
今度はジメドン同士、
分子間の水素結合の形成が重要な因子になってきます。
勉強家の人の中には、「ジメドンはクロロホルム中でケト:エノール比が2:1」といったことが書いてあるのを読んだことがある人もいるでしょう。
でも先の表ではクロロホルム中で、この比は1ケタ違って 20:1 です!
2:1 というのは誤植でしょうか?
実は 2:1 が間違っているわけでもないのです。
表の平衡定数は希薄な状態(< 0.01 mol/L)のもので、
2:1 は濃厚溶液の話なのです。
その昔、H-NMR の感度が低かった時代(まだ「低磁場シフト」といった言葉がリアルだったころ)、
10 % ぐらいの濃厚溶液で測定が行われエノール型の濃度が高く出ていたのを、
今もそのまま、あちこちに書いてあるという次第。
実際に学生諸君の試料の H-NMR を見た時、
溶媒にCDCl3を使っているわけですが、
エノール型のシグナルがはっきり出ているものもあれば、
不純物程度にしか出ていないのもあります
(ここをはっきりさせるために、
スペクトル試料用に再結晶操作を入れるようにしたのです)。
エノール型をはっきり出したいのであれば、
少し試料濃度を高めに調製するのがよいでしょう。
なお溶媒にDMSO-d6 を使うと、
今度はケト型がほとんど見えなくなります。
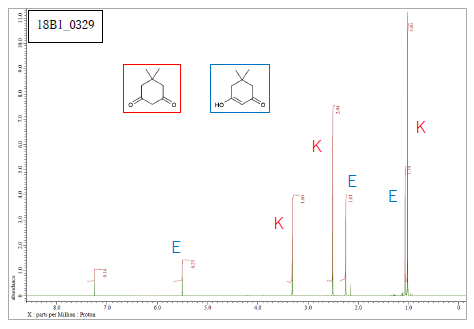
|

|
ジメドン濃度が高い試料(左)と低い試料(右)の H-NMR(CDCl3。600 MHz)
濃度が2倍ほど違うが、
濃度が高くなるとエノール型の比率が大きくなることがわかる
(7.26 ppm のピークはCHCl3、2.16 ppm のピークはアセトン)。
|
表紙のページへ