
水中に沈んだ IRA 400 イオン交換樹脂。
陰イオン交換樹脂を用いて亜鉛を吸着・溶離して分離した後、 オキシン(8-キノリノール)を加えて亜鉛を沈殿させ、重量分析により定量する。 重量分析は亜鉛の分離から始めて2~3日は必要である。 ガラスフィルターの恒量化を他の実験の間に済ませておくとよい。 時間が不足した場合はイオン交換樹脂で亜鉛を分離した後、容量分析で定量する。
(注2)作成したカラムに気泡が混入しないように液面が樹脂の面より下にこない状態で使用する。気泡のまわりでは液体の流れが滞り、分離が不十分になる。
(注3)特に新しいイオン交換樹脂を使用する場合には気体が発生したり、著しく膨潤して流量が極端に落ちたりすることがある。 このような場合は一端ビーカーにイオン交換樹脂を取り、イオン交換樹脂が浸る程度に水酸化ナトリウム溶液を加えてかき混ぜ、 ある程度OH型にしてから、カラムに詰めるようにするとよい。
【亜鉛の吸着・溶離と重量分析による定量】
イオン交換樹脂を用いて亜鉛を分離後、亜鉛を含む溶液にオキシン(8-キノリノール)を加えて亜鉛を沈殿させ、重量分析により定量する。
★これらの操作は2種類の試料溶液について1回ずつ行えばよい。
(注2)放冷時間をできるだけ一定に保つこと。放射温度計でガラスフィルターの温度をチェックしておくのも有効である。
(注3)試料溶液が余分にある場合は溶液量を30 mLに増やしてもよい。
(注4)沈殿は乾燥により無水塩(Zn(C9H6NO)2、式量353.69)に変化する。色の変化に注意せよ。
【亜鉛の吸着・溶離と容量分析による定量】
(注2)赤みが完全に消え黄色くなったところが終点である。終点の判定に不安を感じる者は、6-3の亜鉛溶液を用いて練習実験をしておくと良い。
イオン交換樹脂はポリスチレンなどからなる高分子鎖をスルホン基 (-SO3-)、4級アンモニウム基 (-NR3+) など電荷を持った基で修飾したものである。 こうした高分子鎖は電荷密度が高く、溶液中に溶け込んだイオンを強く吸着する。 またイオンとの親和性はイオンの種類によって異なり、この性質はイオンクロマトグラフィーに利用される。 (一般に電荷の大きなイオンほど、また電荷が同じ場合にはサイズの大きなイオンほど親和性が高い。 たとえば Cl 型の陰イオン交換樹脂に硝酸イオンを含む溶液を流すと、硝酸イオンは塩化物イオンと定量的に交換される。)
亜鉛イオンは塩化物イオンの濃度が高いときにはクロロ錯体([ZnCl4]2- など)を作って陰イオンとして溶存している。 今回の実験ではこのことを利用して亜鉛の吸着・溶離を行っている。 なお、銅もクロロ錯体を形成するが、クロロ錯体が不安定なため塩化物濃度が8 mol/L程度ないと溶出してしまう。 (このことを利用して、銅、亜鉛、ニッケルそれぞれを分離することもできる。最初8 mol/L塩酸でカラムを洗浄してニッケルを、次いで2 mol/L塩酸で銅を、 最後に0.1 mol/L硝酸で亜鉛をそれぞれ溶離すればよい。高濃度の塩酸を使用するので推奨しない)
亜鉛の定量操作については、以前の合金の分析ノート でも紹介しました。 その後の大きな変更点は、分離操作でイオン交換樹脂の量を減らし、ビュレットではなくクロマト管を使うようにした(2009年の生化実験の改定にともなう)こと、 鉄の重量分析(モール塩を硝酸で酸化、アンモニアで水酸化鉄(III)を沈殿分離し酸化鉄(III)として定量)の中止に関わって、 2005年度から亜鉛のオキシン塩による重量分析を正規の課題に組み込んだことでしょうか。 ここではイオン交換樹脂による分離法の実際などについて、 少し詳しく触れておきましょう。
ここで使用するイオン交換樹脂は、 強塩基性イオン交換樹脂の IRA 400 です (アンバーライト® という名前で知られるイオン交換樹脂のシリーズの一つ(現在はデュポン社が製造。日本ではオルガノが販売))。 右図のようなコハク(amber)色の直径 0.7 mm程度のビーズの形で市販されています (最近のIRA 400J は、買いたては淡い黄色ですが、何度も使っているうちにコハク色になってきます)。 見た目は寿司ネタのとびこ(トビウオの卵)をイメージしてもらえばよいでしょう。 亜鉛イオン Zn2+ を分離するのに、 陰イオン交換樹脂を使用するのはちょっと不思議ですが、 塩化物イオンが多量に存在する(> ~1 mol/L)条件下では、 亜鉛がクロロ錯体を作る([ZnCl4]2-など)ことを利用するわけです。
イオン交換樹脂を使用するにあたってのポイントは、 ともかくよく洗うことでしょう。 そもそもが中心となる分離操作が、 イオン交換樹脂に試料を吸着させた後、 吸着されない金属イオンを洗い流し、 洗い終わったら今度は条件を変えて、 吸着された亜鉛を洗い流すという、 ひたすら洗うものなのですが、 それ以外にも「洗い」には気を配る必要があります。
まずはイオン交換樹脂のビーズが残らないように、使用器具や手をよく洗います。 よくある話ですが、イオン交換樹脂のビーズがクロマト管やビュレットの先に詰まって、 液が出なくなってしまうことがあります。 洗い残りのイオン交換樹脂のビーズが1粒でも残っていると、 これが悪さをするのです。 特にアルカリで処理した後のイオン交換樹脂は、 ガラスなどとの付着力が強くなっているので注意します。
綿くずや潰れたイオン交換樹脂のビーズの破片などを、 最初によく洗って取り除くのも重要です。 イオン交換樹脂は再利用して使用するので、 先輩たちが残した綿くずなどのゴミが入っていたりします。 特に細かいビーズの破片が入っていると、 これがカラムに詰まって、液の流れがとても悪くなることがあります。 何度かかき混ぜて、 沈みにくい微細なイオン交換樹脂の破片などを取り除きます。 あまりもったいないなどと思わずに、 屑ビーズを大胆に取り除いた方が、 後の操作がスムーズなようです。 以前は(50年以上前?)こうした操作は「選別 classification」としてかなり念入りに行われたものですが (ハミルトンの水流法などは有名)、 市販のイオン交換樹脂の質が向上して、 そんなに気合を入れなくても大丈夫なようです(さらに昔は「粉砕」をしていたらしい)。 なお全体に言えることですが、操作中にガラス棒などでゴリゴリ強くかき混ぜて、 ビーズを潰さないように注意します。
なお JIS では使用するイオン交換樹脂の粒径としてもっと細かいもの(JIS H 1062 では0.18~0.25 mm)を指定していますが、 流速が稼げず時間がかかるので、通常市販されている粒径 0.55~0.75 mm のものをそのまま使用しています。 吸着が十分行われるのか不安なところもありますが、 分析結果を見る限り、問題はありません (イオン交換樹脂 IRA 400の交換容量は 1.4 meq/mL 以上ということになっていて、 試料溶液 20 mL に含まれる亜鉛の量(0.2 mmol 程度)の数十倍の吸収能があります)。

|
イオン交換樹脂カラム。
内径 8 mmのパイレックス管(さる筋からの頂き物)の先をバーナーを使って引き延ばし、
内径 1~2 mm程度のシリコンチューブを付けてある。
内径 8 mm なので、断面積はほぼ 0.5 cm2。
高さ 2 cm が 1 mL に相当し、イオン交換樹脂 10 mL は 20 cmの高さになる。
チューブの液の流れの開閉によくあるピンチコックを使ってもいいが、 強めのクリップを使うのがお手軽で便利。 |

|
イオン交換樹脂の懸濁液。 メートルグラスに入れ沈んだところで、 だいたい 10 mL の嵩があればよい。 洗瓶で水をかけ、洗い落とす感じで、 クロマト管に入れていく。 |
ここで使用するイオン交換樹脂カラムは、 外径 10 mm 内径 8 mm の先端を少し引き延ばしたガラス管(クロマト管と呼びます)に、 イオン交換樹脂を詰めて作成します。 最初に脱脂綿を少し取って詰めておき(5 mm ~ 1 cm 程度の厚み)、 イオン交換樹脂の懸濁液を流し込んで作ります。 ガラス管がなければ 25 mL のビュレットを流用するのも可です (JIS H1211 や H1062 では25 mL のビュレットを使用することになっています)。
最初に脱脂綿を詰めておくわけですが、 針金やガラス棒でギュウギュウ詰めるのは流速が落ちるのでよくありません。 脱脂綿を指先で丸めたものを、水で洗い落とすぐらいでいいでしょう。 脱脂綿の量は難しいところです。 多すぎると流速が落ちて時間がかかり、脱脂綿に付着した液を洗い落とすのが厄介になります。 少ないとイオン交換樹脂のビーズが漏れ落ちて、ガラス管の先端のくちばしに詰まって液が出てこなくなることがあります。 脱脂綿の量が「必要最小限」の場合、 液を流しているうちにクロマト管のすぼまったところに詰まって、圧縮され断面積が小さくなり、 かえって流速が落ちるケースもあるので、 ちょっと多めにしておくのが無難です。
クロマト管の先の方に綿を詰めた後、 イオン交換樹脂の懸濁液を流し込んで詰めます。 流し込む時に途中で液切れして、気泡が入らないように注意します。 一端気泡が入ると、除くのはなかなか難しい。 また流し込むのにあまり濃厚な懸濁液にすると途中で詰まったりするので、 少し気長に構えて薄めの懸濁液になるようにします。 ここらへんの”呼吸”はなかなか難しい。 こうした懸濁液の挙動は、 実生活でも重要で(大きいところでは土石流、あるいは生コンなどイメージしてもらってもいいでしょう)多くの研究がなされてきたところですが、 懸濁粒子の個性、濃度、流速は元より、どのように懸濁させたかといった履歴にも依存し、 理論的にも極めて難しい問題を多く孕んでいます。
イオン交換樹脂を詰めた後、 その上に脱脂綿を入れることも行われます。 こうすることで、液を注入した時にイオン交換樹脂が舞い上がったり、 あるいは密度の高い液の場合にイオン交換樹脂が浮かんでくる ことを防ぐことができますが、 さして必要ありません。
イオン交換樹脂を流し込んだら、水の流下速度をチェックしておくのがいいでしょう。 シリコンチューブを全開にして、10 mL/min 程度(10 秒で 3 cm 水面が低下)あれば何とかなります。 この段階で流速が 10 mL/min を下回ると、 気泡が入っていたり、何か詰まっている可能性があります。
先のイオン交換樹脂を詰める操作でも触れましたが、 イオン交換樹脂のカラムに液を流す時には、 カラムに気泡が入らないようにしないといけません。 ぼんやり流していると、イオン交換樹脂の上に注いだ液がなくなって、 カラムから液が抜け空気が入ってきます。 気泡が入ると気泡の周辺では流れが淀むことになり、 洗浄が不十分になります。 また流路が狭まり、流下速度が落ちます。
気泡が浮力で浮かんでくる、あるいは流れに乗って押し出されてくれればいいのですが、 表面張力が働くので簡単にはいきません。 気泡を除くにはカラムを上下方向に振動させるのが有効ですが、 下手に振るとかえって事態を悪くします。 いっそ全部取り出して、イオン交換樹脂を詰め替えるのが近道なことが多いです (全量詰め替えを明示した JIS 規格もあります(JIS M8124「鉱石中の亜鉛定量方法」))。
カラムに気泡を入れないためには、 ともかく液切れを起こさないことです。 カラムの液がなくなってきたら、 液を継ぎ足していきます (この意味でクロマト管の長さは 40 cm ぐらい欲しい。イオン交換樹脂が 20 cm + 液を溜める部分が 20 cm、それぞれ 10 mL)。 今回のイオン交換樹脂による分離作業では、 1時間くらいは、ひたすら液を流し続けることになります。 この間、心の空白が生じないようにするのが肝要です。 なお実験によっては、この液を流す作業が数時間に及ぶこともあり、 昔からあの手この手、いろんな試みがなされてきたところでもあります。

|

|
| カラムに気泡が入り、 何とか気泡を取るべく苦闘しているところ。 うまくいったら儲けもの。 詰めなおした方が速いことも多い・・・ | サイホンを使ってカラムに液を供給する工夫。 ビーカーに入った液をシリコンチューブで、 カラムの上方の液だまりに供給する。 |
ここで使うイオン交換樹脂 IRA 400 は、ポリスチレンの高分子鎖に、 4級アミノ基(-N+(CH3)4 Cl- ) を付けたものです。 最初どんなイオンが付いているか知れないので、 使用前に塩酸を通して対アニオンを塩化物イオンに置換して Cl-型にして、 条件を整えます(コンディショニング)。
実験テキストではかなり念入りなコンディショニングを課していて、 まず水酸化ナトリウム溶液で洗うように指示しています。 これはもともと古い JIS 規格にあったものを踏襲しています。 というか、以前(~40 年前?)はこれが当たり前でした。 最初にアルカリで処理して、イオン交換樹脂の製造時に付着している不純物を除き、 水酸化物イオン OH- は中和反応を起こすので、 塩酸で処理すれば容易に Cl-型になるという次第です。 今では市販品の質が向上し、アルカリによる処理は省略されることが多く、 現行の JIS H1062「銅及び銅合金中の亜鉛定量方法」では、 単に 2 mol/L の塩酸を通すだけになっています (JIS H1356「アルミニウム及びアルミニウム合金中の亜鉛定量方法」には、 水酸化ナトリウムによる処理が残っています)。 実験で使用してもらうのは再生品ということになりますが、 この合金の課題以外には使用していないものなので、 これに準じてアルカリ処理を省いてよいでしょう。 ただ何かのまちがい(これがありえるのが学生実験)で亜鉛が吸着されていると問題。 またアルカリ処理をすることで、 イオン交換樹脂の様子が目に見えて変化するのは経験しておいてよいことかと思い、今も残してあります (以前は水酸化ナトリウム溶液の濃度はさらに高く設定して 4mol/L にしていました)。 もし時間がなければ、水酸化ナトリウム溶液による処理はスキップしてもいいでしょう (イオン交換水での洗浄は忘れずに)。
なお水酸化ナトリウム溶液を調製する時、発熱して熱くなりますが、 熱いまま流すとイオン交換樹脂の分解が起きるので、 冷ましてから流すようにします。 また水酸化ナトリウム溶液を流すと、 イオン交換樹脂が OH 型になり、色が赤黒い感じになって膨潤します。 特に新品の場合には膨潤が大きく、 気体が発生したりすることもあります。 こういう場合にはカラムで流しながらコンディショニングするのではなく、 ビーカーに取って、バッチ方式で水酸化ナトリウム溶液と混合、 デカンテーションで数回洗浄して塩酸で中和した後 (OH 型だとガラスなどとの付着力が大きく扱いが面倒)、 カラムに詰めるという手順を踏んだ方がよいでしょう。
イオン交換樹脂は、一般にサイズの大きいイオンの方を強く吸着する性質があります (このあたりの理屈は深く追求しない・・・)。 ですから塩化物イオンよりは亜鉛のクロロ錯体([ZnCl4]2- など)や 硝酸イオン NO3-などの方が強く吸着されます (カチオンでも同様で、たとえば一般に土壌はナトリウム Na+ より、カリウム K+、セシウム Cs+ などの方を強く吸着する)。 このことを利用して、実験ではまず 2 mol/L 塩酸を加えて塩化物イオンを高めた状態で、 試料溶液中の亜鉛をクロロ錯体の形でイオン交換樹脂に吸着させ、 その後、塩酸で洗浄することで銅やニッケルを除いて亜鉛を分離します。
テキストにもあるように塩化物イオンの濃度が 2 mol/L 程度であれば、 ニッケルや銅のクロロ錯体は不安定で吸着されません。 ただし今回は試料中の鉄の含有量が少なく無視できるのですが、 仮に鉄(III) Fe3+ が含まれていると [FeCl4]- などが生成して、 一緒に吸着されてしまいます。 そこで鉄を含む試料については、塩酸にアスコルビン酸(ビタミン C)を加えて鉄(II) Fe2+ に還元するという処方が取られます。
亜鉛をクロロ錯体の形で吸着させた後、 取り除く(溶離する)には、いくつか流儀がありますが、 ここでは希硝酸を使う処方を採用しています (アンモニア-塩化アンモニウム溶液を使用する処方はちょっと臭うので敬遠)。 塩化物イオンの濃度が低下し、亜鉛のクロロ錯体が分解すれば、 おのずと亜鉛は出てくるというわけです。 なおイオン交換樹脂の交換容量が 14 meq 程度(1.4 meq/mL × 10 mL)なので、 硝酸をちょうど当量程度(15 mmol = 0.1 mol/L × 150 mL)流して、 最終、イオン交換樹脂がほぼ -NO3 型になるぐらいに仕込んであります。
このイオン交換樹脂の操作で、2 mol/L 塩酸を多用しているのを疑問に思う人もいるでしょう。 塩化物イオンの濃度が問題なら、学生実験でもあり、 もっと安全、安価な塩化ナトリウム NaCl 溶液を使った方がいいはずです。 やってみたことはありませんが、おそらく塩化ナトリウム溶液でも実験はできるでしょう。 けれども塩酸の方が実験はスムーズに進むはずで、 実際にこうした用途では、たいていの場合塩酸が使用されています。
-OH 型を -Cl 型にする上で中和反応を利用できるので効率的、 また塩基性塩の生成を防げるというのは無論ですが、 それだけではありません。 まず粘度と密度を見てみると、20 °C で、2 mol/L の塩酸の粘度は 1.10 mPa s、密度は 1.03 g/cm3。 一方 2 mol/L の塩化ナトリウム溶液の粘度は 1.22 mPa s、密度は 1.08 g/cm3。 塩酸方が 5 %くらい流速がかせげます。 また粘度が小さいことは、イオン交換樹脂のビーズを洗う上で有利です。 さらに重要なのは、塩酸は蒸留して精製できるので、 高純度のもの(JIS 規格で一番多量に許容されているナトリウムで 0.1 ppm 以下)が安価に入手できることです(お値段 ~ 800 円/500 mL(35 %))。 塩化ナトリウムはこの点、普通に手に入る JIS 規格の試薬では十 ppm オーダーのカルシウムやマグネシウムは覚悟しないといけません (お値段 ~ 900 円/500 g。)。
すでに「セスキ炭酸ナトリウム(トロナ)の塩酸への溶解」の課題で、 半定量的なレベル(~1/100 g)でその一端には触れてもらっているのですが、 ここではさらに定量的なレベル(~1/10000 g)での重量分析に挑戦することになります。 実験的に「質量保存の法則」が成立するには、いろいろ困難な事情があることを体得していただければ幸いです。
なお一般に重量分析には時間と忍耐を要します。 今回の場合、亜鉛の分離から始まって最終的な結果を得るまでに2日間は見込む必要があります。 進度が遅い場合には、容量分析を採用することを推奨しています。
オキシン(最近はもっぱら 8-キノリノールと呼ばれていますが、個人的にオキシン oxine という昔ながらのニックネームが気に入っています) がマグネシウム、アルミニウム、亜鉛、鉄、銅等、多くの金属イオンと沈殿を作り、 それが定量に利用できることが知られたのは、もう100年近く前のことでした。 ジメチルグリオキシムのように金属イオンに対する選択性が高くないのですが、 今回のようにすでに分離操作を終えた試料から、 金属イオンを回収するのには便利に使えます。 また後の吸光光度法の課題でも出会いますが、 適当な沈殿剤のあまりないアルミニウムの定量などには欠かせない試薬になっています。 なお pH を調整することで、金属イオンの分離もある程度可能で、 沈殿の pH 条件について古くから研究が行われています(オキシンは2塩基酸で pK1 = 4.9、pK2 = 9.8)。
亜鉛のオキシン塩(オキシン錯体)の沈殿生成の pH 条件は 4.5~13 ぐらいです。 pH を高くして pH 8 を超えると、マグネシウムやカルシウムも沈殿するようになるので、 酢酸緩衝液(pH は 4.5 くらい)で pH を調整しています (下敷きにした JIS H1211「黄銅分析方法」(廃版)では、pH が 4.6 になるように指示されています)。 オキシンを加えるについては流儀があり、ここではオキシンのエタノール溶液を加えるようにしています。 バーナーで加熱したりするところでエタノール溶液を加えるので、 危険と言われればそうなのですが、 これぐらいの緊張感はあった方が個人的にはうれしいです。 エタノール溶液ではなくオキシンの酢酸溶液(1 mol/L ぐらいの酢酸にオキシンを溶かす)を使うのもあって、 これは吸光光度法の課題で採用しています。 今回の場合は、酢酸溶液を加えると pH が 4 ぐらいまで下がり、 pH の調整に一手間かける必要が出てくるので、エタノール溶液にしています(JIS H1211「黄銅分析方法」(廃版)も同様)。
なお 亜鉛 10 mg(0.15 mmol)あたりオキシンは最低で 44 mg(0.3 mmol)必要で、 JIS H1211 では、だいたいこの10倍量、500 mg 程度のオキシン(2.5 %溶液 20 mL)が必要という指定になっていました。 そこで当初はオプション課題であったこともあり、使用する試料溶液を 10 mL、含まれる亜鉛の量はおおざっぱに 10 mg として、 溶離した溶液に2.5 w/v %オキシン溶液を 20 mL 加える処方にしていました。 それを2005年度から正規の課題に移行するにあたってスケールを2倍して、使用する試料溶液を20 mL にした際、 オキシン溶液の量を削り込んだのです (単純に加えるオキシン溶液を2倍にすると、溶液のアルコール濃度が高くなりすぎる)。 試料溶液中の亜鉛量を、元の洋白の亜鉛含量を 30 %とすると、 試料溶液 20 mL 中には亜鉛が 15 mg(0.23 mmol)入っている。 この 10 倍量でオキシンは 4.6 mmol、0.67 g。 2.5 w/v %オキシン溶液にして 27 mL。 というわけで、実験テキストにある 27 mL という、いささか切りのよくない数字が出てきています。

|

|

|

|
| 順調に亜鉛の分離ができておれば、 普通はこんな感じ | イオン交換樹脂による分離が不完全で、銅やニッケルが混じっていると浅黒くなる。 この程度なら無視しても可。 近年、こういうケースに出うことが増えているように感じます。 | ここまでになると、やり直した方がよいでしょう | 冷やして置いておくと、オキシンの針状結晶が析出してくることがあります。 中性条件だとオキシンの溶解度は室温で0.06 mass%ぐらい。 |
重量分析に用いるグラスフィルターは、 足のないルツボ型のものです (足の付いたロート型のものはアダプターが必要なく、 取り扱いが手軽で再結晶などには便利。 ただし乾燥させて重さをはかる分には、足の部分の水分がなかなか抜けなくて不利)。 吸引瓶にアダプターを付けて使用します。 このアダプターを商品カタログなどで「グーチロート」と呼んだりしていますが、 「グーチロート Gooch funnel」というのは海外では通用しません
今ではほとんど使われていないでしょうが、グーチルツボ Gooch crucible というのはあります。 元来のグーチルツボは、ルツボの底に小さい穴をたくさん開けたもので (時に1878年)、 ブフナーロートのルツボ版と思っておいてもらえればいいでしょうか (もっともブフナーロートの方が後輩(1888年)です)。 白金製、磁器製でろ過後、バーナーで強熱するものだったのですが、 ガラス製のものができ、ここで使用するルツボ型のグラスフィルターは、 グーチフィルター Gooch filter と呼ばれたりします。 このアダプターについては適当な名称がなく、単に”アダプター”というだけでは気が利かないので、 「グーチロート」と呼びならわしているようです。

|

|

|

|
| ルツボ型のグラスフィルター。 | グラスフィルターを吸引瓶に接続するアダプター。 日本では「グーチロート」と呼ばれることがある。 | アダプターにゴムのチューブを切ったものを嵌め、 吸引瓶に付ける。 | グラスフィルターをセットしたところ。 後は吸引ろ過。 |
再結晶で結晶を取り出す時には、 99 %も回収できれば良しとしたものです(ここらへんにこだわり始めると神(オタク)の領域か)。 またニッケルジメチルグリオキシムの場合には、 ざっくり懸濁液にしてろ過した後、 ビーカーに付着した沈殿は塩酸で溶解して回収するので、 基本、ビーカーを洗えばよいだけでした。 けれどもここでは沈殿を定量的(1/1000 オーダー)に回収する必要があります。
こうした時の小道具によく使われるのが「ポリスマン」と称される、 ガラス棒にゴムのチューブをかぶせたものです。 溶液をかき混ぜて沈殿を懸濁させて大部分を吸引ろ過した後、 ビーカーに付着した沈殿を、 ポリスマンを使ってこそげ落として懸濁液にし、 吸引しながらグラスフィルターに洗い落とすのです。
このポリスマンを作る時には、 チューブは短めにして水で濡らし ガラス棒はできるだけチューブに近いところを持つようにします。 無理にチューブを嵌めようとすると、 ガラス棒が折れて怪我をすることがあり、 ここ十年ばかり、ガラスによる大きな外傷は、 このポリスマンに関わって起きています。

|

|

|
| ガラス棒と短く切ったシリコンチューブ。 | 「ポリスマン」 | 折れたポリスマンで手をざっくり切った学生が、 後に残していったもの(付いていた血は洗い流してある)。 チューブを長めに取り過ぎて(ガラス棒もちょっと長い)、 嵌めるのに無理をしたらしい。 |
加熱乾燥には、減圧乾燥器(東京理化器械 VOS-301SD)を用います。 グラスフィルターをアダプターから取り外して(手の油などつかないよう、ゴム手袋をして作業するのが望ましい)ビーカーに入れ、 時計皿で蓋をして減圧乾燥器に入れるようにします (減圧乾燥器内部は、 空気の出し入れにともなって風が吹き、 沈殿が吹き飛ばされたり、 ゴミが舞い込んだりすることがあるので注意)。 温度は 140 °C で、ダイアフラムポンプを用いて 1~2 kPa (0.01~0.02 気圧)ぐらいまで減圧します。 実験室には減圧乾燥器が2台あり、 それぞれが 【30 分減圧加熱】 - 【10 分 入れ替え】 - 【30 分減圧加熱】 - 【10 分 入れ替え】 - ・・・ というスケジュールで交互に運転するようにしています。
実験する立場でいうと、【試料を入れ 30分加熱】 - 【取り出して 30分放冷】 - 【秤量】 - 【試料を入れ 30分加熱】 - 【取り出して 30分放冷】 - 【秤量】 - ・・・ という段取りで、1サイクル 1 時間20分程度で回してもらえれば、 減圧乾燥機の 1サイクル40分の運航スケジュールとだいたいかみ合って進みます。 ビーカーの出し入れの際は、いささか熱いので用意してある厚手の軍手を使うのがよいでしょう。 またグラスフィルターを取り出す時は、 熱くてゴム手袋では融たりすることがあるので、 ルツボばさみ(トング)を使います。
この加熱乾燥の間に、得られた亜鉛のオキシン塩の二水塩は無水塩になり、 下の写真にあるように、薄黄色だったものが黄色くなります。 また写真で減圧乾燥機の窓が少し曇っているのは、 洗浄が不十分で残っていたオキシンが昇華して窓に付いているためです (困ったことですが・・・)。

|

|

|
| 減圧乾燥器。 ビーカーにグラスフィルターを入れ、 ゴミが入らないように時計皿で蓋をして入れる。 名前を忘れないこと! | 運転中の減圧乾燥器。 設定温度は 140 °C。 現在減圧中で、最終 1 ~ 2 kPa になる。 | 加熱乾燥前(左)と加熱乾燥後(右)の亜鉛オキシン塩。 無水塩になると黄色味が増す。 |
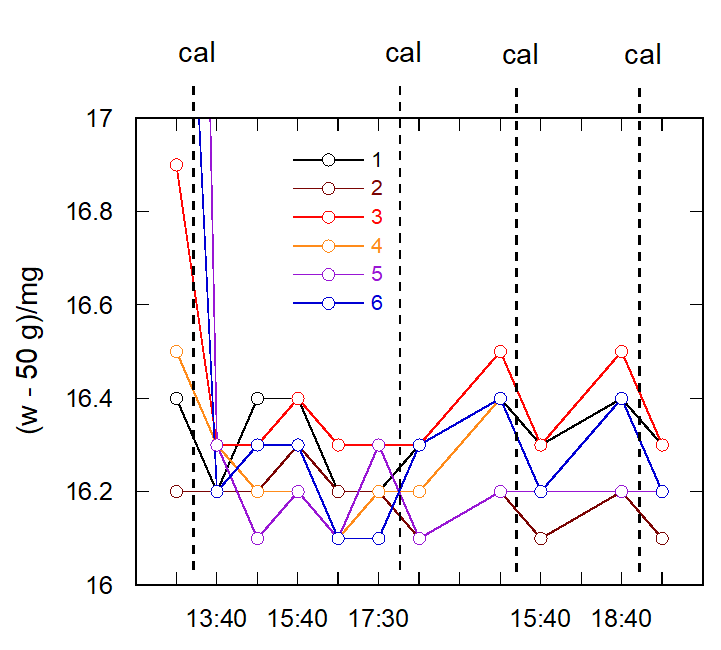
今回の重量分析では、およそ 30 g のグラスフィルター (とで採取したおよそ 60 mg の亜鉛のオキシン塩) の重さのばらつきが、 加熱・冷却を繰り返しても 0.3 mg 以内であることが求められています。 加熱・冷却には1時間程度必要で、したがってその間に天秤の秤量値が 0.3 mg 変動するようだと、 天秤の安定性の実験をしているようなものになるので、 変動は 0.1 mg 程度に収まって欲しいところです。
現在、学生実験では 6 台の精密天秤 (”化学天秤”。典型的には感量が 0.1 mg で最大秤量 100 g ぐらいの天秤。 現在使用している機種は Mettler の ME104) を使用しています。 その6台について、5月中旬、合金の分析の課題を実施しているときに、 50 g の古い分銅を使って各天秤の秤量値の変動を調べた結果を右図に示します。
ME104 は内蔵分銅を備え、手動で較正を行うようになっています。 図に示したのは2日間にわたる結果で、 初日、1か月ぐらい較正をしていない状態の天秤についてまず秤量値を調べると、 案の定、大きく外れている天秤では 4 mg ぐらい値が外れていました。 較正をかけて秤量値を調べると、6 台とも 0.1 mg 以内で一致。 この日はまだ重量分析に入る学生が少なかったので、何度か途中で秤量値のチェックをしてみましたが、 あまり大きな変動はなく、 午後5時30分、実験終了時のチェックで、1台だけ 0.2 mg の減少が見られたほかは、0.1 mg に収まっていました。
この翌日、重量分析に入る学生が増えて、たまたま天秤室が空いた午後3時40分ごろチェックしてみると、 前日の値から 0.2 mg 外れているのが 3 台、較正をかけると 6 台とも前日の結果と 0.1 mg 以内で一致しました。 その後、精密天秤を利用する学生が増え、 最終午後6時40分、実験終了時のチェックで、3 台が 0.2 mg 増加を示し、 較正をかけると、6 台ともこの日の最初に較正をかけた時の秤量値になりました。
これ以外にも以前から何度かチェックしているのですが、 どうも学生の出入りが少ない時は天秤の値は落ち着いていて、 多いときには少し変動が見られるようです。 これは室温の変化によるものでしょう。 ME104 の温度による変動率は 2 ppm/K ということになっていて、 室温が 1 °C 変わると、50 g の分銅であれば 50 g × 2× 10-6 = 0.1 mg の変動が現れます。 天秤室に人が多数入って、実験の間に室温が 2~ 3 °C 上がったとすると、 だいたい変動は説明できます。
もう一つ注意しないといけないのは、内蔵分銅で較正しても、天秤によって 0.2~0.3 mg 程度、秤量値が異なってくることです。 これは扱っているのが 50 g ぐらいの物体で最大秤量(ME 104 では120 g)の半分ぐらい。 機種による直線性からの外れの違いが顕著に出てくることに由来するようです。 ME104 では直線性が 0.2 mg ということになっていて、 天秤ごとの差が最大 0.3 mg は、そもそもの天秤の仕様通りということになるでしょう。
学生実験の場合、大人数の実験なので、どうしたところで室温の変化は避けられません。 室温の変化の影響は天秤の較正を行うことで、ある程度回避できますが、 較正の前後で秤量値が不連続的に変動すると、(均してみると正確になるわけですが、) 較正のタイミングによっては、一部の学生に”不利”に働くことになりかねません。 そこで通常は、重量分析の当日、実験開始前に較正をかけるだけにしています。 一方、天秤による直線性の違いは、同じ天秤を使って秤量すれば、回避可能です。 というわけで、実験にあたっては6台ある中のどれか同じ天秤を使うよう指示しています。

|

|
| 天秤室の様子。 6台並んだ除振台の上に精密天秤が置いてある。 | 天秤にチェック用の古い 50 g の分銅を置く。 古くなると少し錆びるためか、若干重くなる。 |
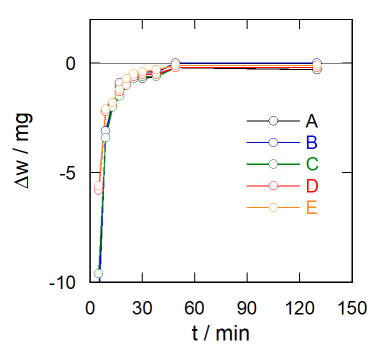
ここでの重量分析は、グラスフィルターの空の時と、 沈殿を採取した時との重さの差を求めるという、 いたってシンプルなものです。 けれども求めるのが30 g の物体の 0.1 mg オーダーの差、 十万分の1 以下、百万分の1 の世界の話なので、 慎重な配慮が求められます。 いくつかポイントがありますが、 まず安定に秤量できるまで冷ますことです。
グラスフィルターは、デシケーターに入れて放冷します。 デシケーターに入れる時は、ルツボばさみを使って、 冷めやすいようにビーカーから取り出して、 グラスフィルターをデシケーターの陶板の上に置くようにします。
温かいものをそのまま空気中で天秤に載せて秤量すると、 空気の対流が起きて軽めにでます (このことはビーカーなりを手で温めるぐらいでも確かめることができます)。 右図に示すのはバーナーで加熱した磁器ルツボ(重さ 6 g程度)の秤量値が、 放冷とともに変化するようすを示したものです。 熱いうちは数mg オーダーで小さい値になっているのですが、 だいたい30分ぐらいたつと、秤量値は落ち着いてきます。 ただし完全に落ち着いているわけではなく、 さらに放置すると、そこから 0.5 mg程度増加します。 グラスフィルターは図に示すルツボの数倍重いのですが、 140 °C までの加熱ということもあって、やはり30 分ぐらいたつと秤量値は落ち着いてきます。
完全に冷めて、対流の効果がなくなるまで待って、 重さをはかるのが望ましいのかもしれませんが、 それには1時間以上必要です。 そこで一定時間後、秤量値の変化が秤量の間に認められなくなる程度まで放冷して、 それを恒量値と見なす方針を取ります。 ここでは30分程度ということで、やってもらっています。 ですから帰宅間際まで加熱し、翌日に秤量という手順を踏むと、 放冷時間が長くなって 1 mg 近く重く出てきたりして、無駄足を踏むことになってしまいます。
恒量化操作では、【30分加熱】 - 【30分放冷】 - 【秤量】という操作を繰り返すことになります。 この1回のサイクルで「恒量」になったかどうかを知るには、 もう一度このサイクルを行って、秤量値が 0.3 mg 以内で一致することを確認する必要があります。 ですから恒量化には最低でも3時間ぐらいかかることになります。 これを1つの試料について、 空のグラスフィルターと沈殿を採取したグラスフィルター、それぞれ行うわけですから、 うまく並行して操作を運んでも、学生実験では2日かかるという寸法です。 2回で恒量に達しなければ、またさらに1サイクル。 ですからグラスフィルターや沈殿の洗浄などを確実に行っていないと後が大変。 細心の注意と時間と忍耐。 これがこの重量分析で求められることと言っていいかもしれません。

|

|

|
| 実験で使用するデシケーター。 300 °C ぐらいの高温のルツボを使用することもあるので、 こうしたすり合わせの蓋を持つ、 ガラス製のデシケーターを使用するのが一般。 | デシケーターに入れるときは速く冷えるように、 グラスフィルターを加熱の時に使ったビーカーから出し、 直接、陶製の板の上に置く。 | デシケーターを持ち運ぶときには、 身と蓋を一緒に持つようにする。 いろいろ流儀があるが、図は石橋先生お薦めの持ち方(石橋雅義「定量分析実験法」)。 |
亜鉛の容量分析では、EDTA によるキレート滴定を行います。 ここでは、金属指示薬としてXO(キシレノールオレンジ)を使用しています。
XO(キシレノールオレンジ)指示薬は、 pH 指示薬に EDTA のようなイミノジ酢酸基(-N(CH2COOH)2)を導入するという設計思想のもとに、 合成された一連の金属指示薬の一つです (クレゾールレッドにホルムアルデヒドとイミノジ酢酸でマンニッヒ反応を起こさせて合成)。 類縁の金属指示薬にカルシウムの蛍光指示薬として有名なカルセイン calcein(これはフルオレセインにマンニッヒ反応をさせたもの)などがあります。
XO を金属指示薬として使用できる pH 領域は 6 以下で、 これ以上になると XO 自体が塩基型になって赤みがかってきます。 また pH 5 以下では亜鉛とのキレートが不安定になってくるので、 pH は 5 ~ 6 にしておく必要があります。 テキストの実験手順で pH の設定にかなり配慮しているのはこのためです。
なお XO は、銅 Cu、ニッケル Ni と安定なキレートを作り、 イオン交換樹脂による分離が不完全で、銅やニッケルが入っていると、 変色が明瞭でなくなります(ブロッキング。このためニッケルのキレート滴定は逆滴定を用いていましたね)。 アンモニアで中和する段階で、 銅アンミン錯体の青紫色が認められたら、 チオ硫酸ナトリウム溶液を加えてマスクするようにします (ニッケルはあったとしてもたぶん少量なので無視。 マスクするのにシアン化カリなどを使用することになり、 そもそものイオン交換樹脂による分離をきちんとこなすのが重要でしょう)。
最後に重量分析と容量分析の精度についてですが、 実験法 II の問題集で取り上げているのは、 まさに学生実験で得られた結果です。 それをどう見るかは人それぞれですが、 ぼくは「学生実験の結果は数字だけではない」と考えています・・・

|

|

|
| XO 指示薬を加えると赤くなる | EDTA で滴定していくと、赤みが消えていく。 右側のビーカーは滴定終了後。 | 滴定終了後の溶液。 |